中晚期非小细胞肺癌首创性药物索托拉西的艰辛历程:让KRAS“不可成药”靶点魔咒成为历史
发布日期:2022-02-12 浏览次数:
 2021年5月28日,美国FDA根据索托拉西临床II期的安全有效性结果,批准其上市,索托拉西成为治疗携带KRASG12C变异的中晚期非小细胞肺癌的首创性药物,引起业界瞩目。KRAS是一种致癌基因,蛋白Gly12容易发生变异Cys12,大约40年前科学家就已揭示KRAS变异与肿瘤的关系,但多年来研究进展缓慢。安进(Amgen)迎难而上,基于片段的药物设计(FBDD)方法和共价结合(Covalent Binding)原理,将药物共价结合于Cys12上,锁定KRAS为不可逆的非活化构象状态,突破了KRAS非可药性靶标的思想束缚,开发出全球首创的以KRASG12C为靶标的小分子靶向药物——索托拉西。
2021年5月28日,美国FDA根据索托拉西临床II期的安全有效性结果,批准其上市,索托拉西成为治疗携带KRASG12C变异的中晚期非小细胞肺癌的首创性药物,引起业界瞩目。KRAS是一种致癌基因,蛋白Gly12容易发生变异Cys12,大约40年前科学家就已揭示KRAS变异与肿瘤的关系,但多年来研究进展缓慢。安进(Amgen)迎难而上,基于片段的药物设计(FBDD)方法和共价结合(Covalent Binding)原理,将药物共价结合于Cys12上,锁定KRAS为不可逆的非活化构象状态,突破了KRAS非可药性靶标的思想束缚,开发出全球首创的以KRASG12C为靶标的小分子靶向药物——索托拉西。
1 研发背景
30%的肿瘤是由于癌基因KRAS突变引发的,是肺、结肠和胰腺癌等常见的肿瘤驱动基因,尤其在肺癌中,20%的KRAS蛋白Gly12变异成Cys12(KRASG12C)。研发KRAS蛋白突变与癌症的相关性已有三十多年之久,但学界和企业界一直没有研制出药物,主要原因是成药性差。一是因为KRAS与GDP和GTP酶的结合力非常强,外来的抑制剂很难达到与GDP和GTP酶发生竞争性结合的强度;二是KRAS蛋白的结构缺乏其他的疏水腔穴,缺乏设计抑制剂的抓手。
为了能找到可结合KRAS中蛋白Cys12的苗头化合物,Ostrem等基于片段药物设计并筛选和评价了380个二硫键衍生物对KRAS-GDP标签性结合力,用质谱检测全蛋白的MS信号,发现化合物2E07(1)和6H05(2)具有较强的结合能力(结合能力:6H05>2E07)。 故以6H05(2)为起始物进行先导结构的演化,得到活性更强的化合物3。
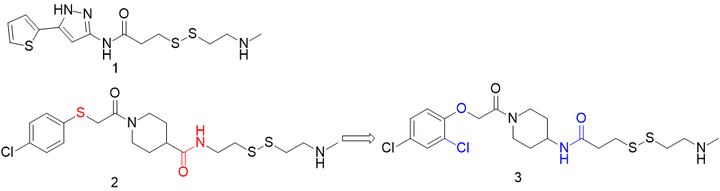
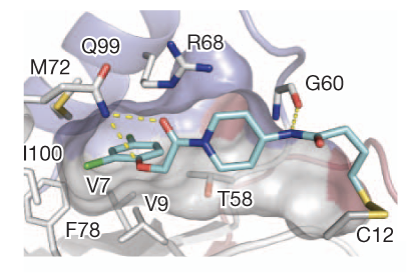
通过化合物3与KRASG12C共晶结构发现,3没有结合在核苷酸结合腔中,而是在变构区的疏水性沟槽上,即KRAS-GDP变构区域的S-IIP处,将KRAS构象固定在非活化状态,并延伸到Cys12以外的区域中,3的胺基与Gly60羰基形成氢键。
二硫键是较弱的亲电性基团,为提高抑制活性,将二硫结构优化为亲电性更强的丙烯酰胺和乙烯基磺酰胺,这类衍生物都呈现活性,但乙烯基磺酰胺的亲电性强于丙烯酰胺而导致选择性差,因而优化了丙烯酰胺系列。其中化合物4是以哌嗪为连接基,LC-MS-MS确证与KRAS的Cys12发生了共价结合,经生化方法测定发现,化合物4抑制KRASG12C的速率常数{kobs/[I]=0.1(mol·L-1)-1·s-1)}显示出一定的活性。进一步优化活性和过膜性,得到化合物5,活性和成药性均得到显著改善。
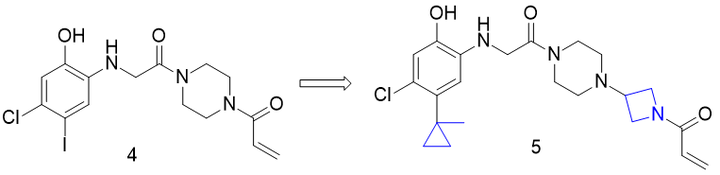
2 先导化合物的发现
安进为了加快研究KRAS抑制剂步伐,与Carmot Therapeutics公司合作,利用Carmet的化学型演化(chemotype evolution)技术平台,设计合成了20000多个单体分子,并使用生化和质谱方法评价了化合物的结合性能,从而得到新的苗头化合物6。
2.1 母核的固定
随后将酰胺基与溴代苯酚环合为吲哚以固定母核的构象,作为探索优化先导物的路径,结果表明,变换吲哚3-位连接的末端片段对活性有一定的影响,但不显著,其中化合物7的抑制活性最好{Cell pERKc 4h: IC50=11.4 μmol·L-1}。
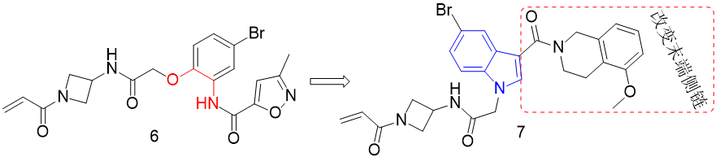
2.2 化合物7结构优化
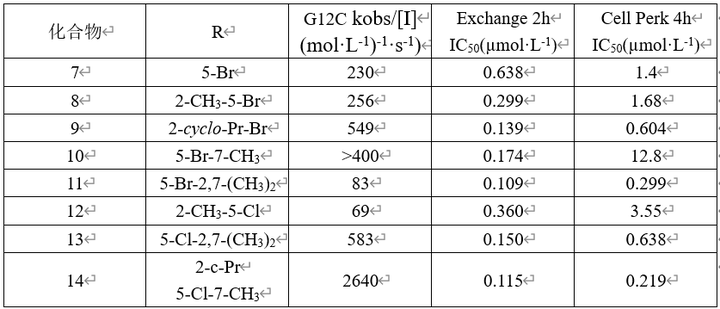
化合物14对细胞抑制活性最好,但是对啮齿类的口服生物利用度较低,体内的清除率却很高,故仍需作进一步优化。
3 先导化合物结构优化
3.1 骨架融合
由于化合物14是经过各片段优化而得到的,要想进一步提高其成药性,只能另辟蹊径。科学家从分析化合物与KRASG12C的结合模式入手,发现化合物14和15都是定位于KRASG12C的变构区S-II区域,通过叠合化合物14和15 的活性构象,发现在化合物14的N1位连接出疏水片段,相当于15的四氢异喹啉所占的位置,因而合成了以酞嗪为母核的新类型化合物。
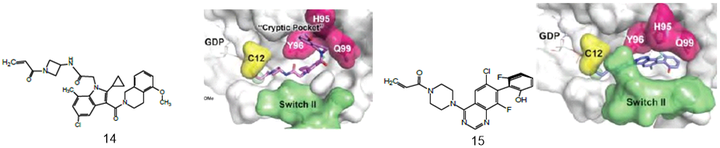
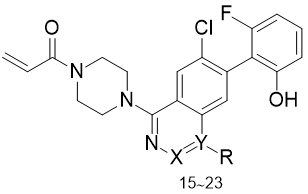
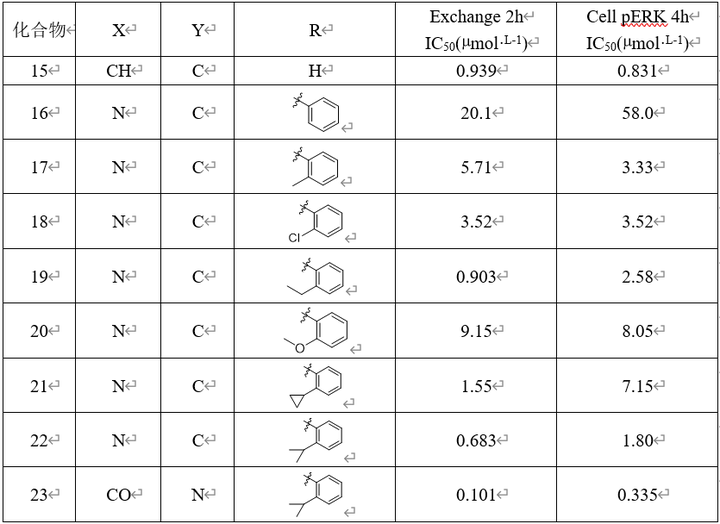
相较于化合物15,化合物23生化和细胞活性分别提高了3和9倍。共晶结构还显示化合物23的喹唑啉酮环与异丙基苯环的构象呈垂直取向,由于邻位的双取代造成的阻转异构,存在光学异构体,经色谱拆分和测定活性发现R-23为优映体,抑制细胞活性的IC50=0.130 μmol·L-1,显著高于S-异构体。
3.2 结构优化
进一步通过在喹唑啉酮的7位引入大疏水基团、在哌嗪环的2′位作甲基取代和母核8位元素CH作N等排变换对活性的影响。
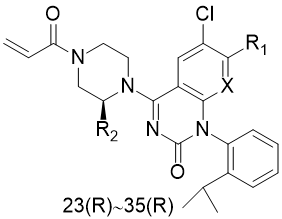
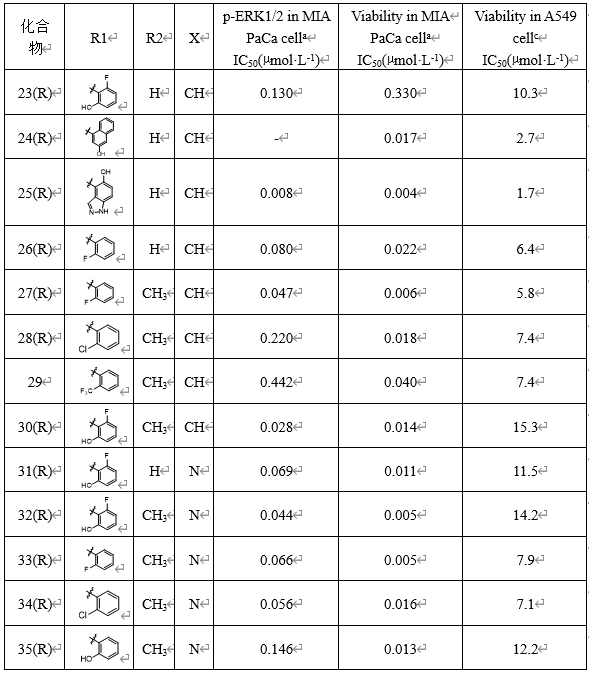
化合物R-32的过膜性显著提高,而且溶解度也提高了,生物利用度达到33%。将R-32的取代基变换为氟、氯、羟基,活性和药代性质的综合效应都不如R-32,提示结构的优化需要在活性和成药性之间作调整。
3.3 解决阻转异构的困惑
化合物32的光活体在25℃逐渐消旋化,半衰期为8天,转化自由能垒∆G≠=26 kcal·mol-1,所以这对于将R-32研发成药物是个障碍。解决的办法是变换结构,途径有3个:(1)提高构型转化的能垒,使活化自由能ΔG≠提高到30 kcal·mol-1,这样在室温下能以稳定的构型存在;(2)将转化能垒降低到20 kcal·mol-1,成为容易转化的混合物,研发成消旋体;(3)制备对称的取代基,消除手性轴。为此,采用两种方法研究具有阻转异构的化合物转换构型的能垒。设计合成了一系列衍生物并测定活性。
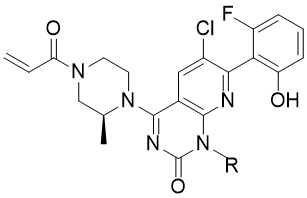
化合物46、47和48取代的对称性化合物活性都很强,而且也阻止了手性键的旋转。
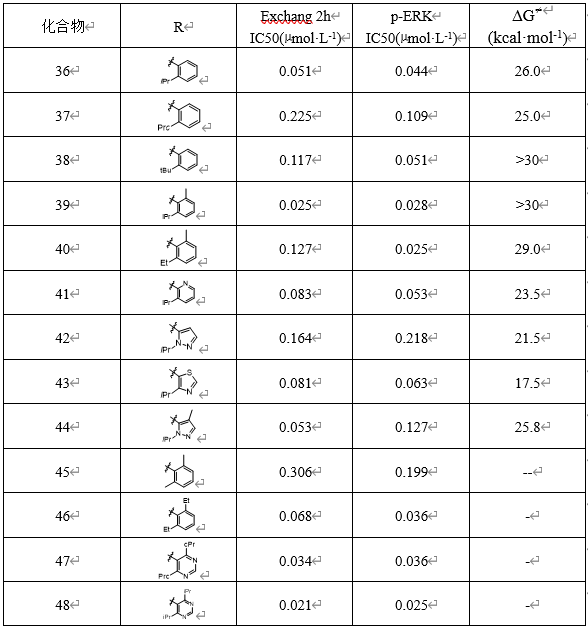
3.4 高活性化合物的体内活性
对活性较好的5个化合物(36、43、46、47、48)进行药动学评价,它们的半衰期、清除率和血浆蛋白结合率(或表征为游离的化合物的份额)相差不多,但对小鼠的口服生物利用度(F)不同,36与43的F值相近,而其他3个的吸收性差。化合物36小鼠灌胃10mg·kg-1的最大血药浓度(Cmax)是IC50的4.5倍,43为1.5倍,提示36的体内活性强于43。所以优选出36是高活性化合物中药代性质优良的化合物。
3.5 生物药剂学性质的优化
化合物36的胜出是将苯并环换作吡啶并环,提高了该片段与“神秘”腔穴的结合力,为了提高其物化和生物药剂学性质,对吡啶并环的氯与氟原子进行变换,苯环用吡啶环变换,合成的化合物结构与性质评定如下表。
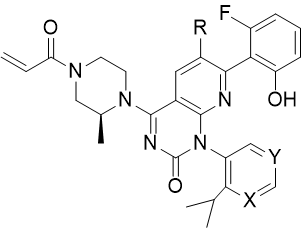
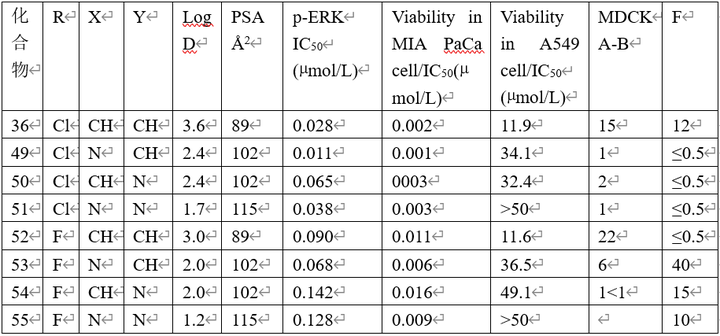
结果表明,化合物53~55的溶解性都提高,53的细胞活性略有降低,但过膜性和生物利用度都显著的提高,是个突出的优化分子。
4 候选物的确定
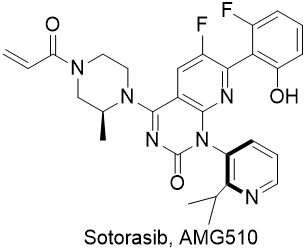
R-53的体内药效学表明,灌胃移植MIA PaCa细胞的裸鼠模型,在10mg·kg-1剂量下显著抑制ERK的磷酸化,进而评价了剂量-时间的药效/药代的关系,以及对动物安全性试验,证明R-53是选择性KRAS抑制剂,经系统的临床前研究后,定名为索托拉西(sotorasib, AMG510)于2018年开始临床研究,历时三年后基于II期开展的名为CodeBreaK100的临床研究,对126例KRASG12C变异的中晚期肺癌在免疫疗法和化疗无效的患者表明应答率为37%,中位应答时间10个月,88%患者症状明显改善,于2021年5月28日经美国FDA批准上市。
5 小结
在非小细胞肺癌的治疗中,EGFR、ALK、ROS1等靶点早已被一一攻克,只有KRAS基因让人束手无策。KRAS药物研发艰难进展挫败了很多科学家,有人甚至认为KRAS根本无法成为药物的靶点,存在KRAS突变的患者无药可医!但安进迎难而上,打破了KRAS靶向药物的难成药性问题,使索托拉西成为了这一领域的首创药物,让人类终于在KRAS的铜墙铁壁上凿出了一道光。
印度肿瘤药房(India Pharmacy)是印度新德里肿瘤药房信息咨询服务平台,旨在为患者提供各类进口原研 进口仿制 最新研制等医药信息咨询 跨境医药电商直邮服务,让患者轻松获取全球最佳药品有更多选择,基本涵盖新特药 抗癌药 靶向药 丙肝 乙肝 高血压 糖尿病 痛风 等药品,欢迎咨询!官方微信 Yindu7689
